
こちらの記事では、「E2B(R3)実装ガイドに対応した市販後副作用等報告及び治験副作用等報告に関するQ&Aの改正について」例示を用いて説明します。
分かりやすい言葉で説明します。
筆者情報
製薬業界12年以上勤務をしており、今は管理職に就いています。
小さな会社なので、守備範囲も広く、毎日新しいことにチャレンジしてながら、フルタイムで働き2歳と4歳の子育てをしています。
解説する通知
令和2年12月9日
事務連絡
「E2B(R3)実装ガイドに対応した市販後副作用等報告及び 治験副作用等報告に関する Q&A の改正について 」
・医薬品副作用全ての通知はこちらのPMDAサイトからチェック可
副作用等の報告に関する情報
表の見方
【市販後】:市販後副作用報告に関係する内容
【治験】:治験副作用報告に関係する内容
Q&A詳細
(2)報告期限等
| Q16:【市販後】 平成 10 年3月 11 日付け医薬安第 25 号厚生省医薬安全局安全対策課長通知「医薬品の安全対策の徹底について」の記2.(2)において、『添付文書の改訂によって新たに記載されることとなった副作用について、添付文書の改訂が実施され医療機関等への情報伝達が終了するまでの間に当該副作用と同様の情報を入手した場合は、「使用上の注意から予測できない副作用」として取扱い 15 日以内に報告すること。』と規定されているが、医療機関等への情報伝達が終了するまでとは、いつの時点と考えたらよいか? | A16:【市販後】 製造販売業者が行った情報伝達の終了した日又は医薬品安全対策情報[DRUG SAFETY UPDATE(DSU)]が医療機関に配布された日のいずれか早い方の日とすること。 |
補足:医薬品安全対策情報[DRUG SAFETY UPDATE(DSU)]とは?
日本製薬団体連合会(通称:日薬連)が提供する、医療用医薬品添付文書の使用上の注意の改訂情報。他にもPraise-Netを運用していて、医薬品の行政通知や研修(無料も多数あり)をメールでタイムリーに配信してくれます。業界の全員がPraise-Netの会員といっても過言ではないです。
日本製薬団体連合会(通称:日薬連)が提供する、医療用医薬品添付文書の使用上の注意の改訂情報。他にもPraise-Netを運用していて、医薬品の行政通知や研修(無料も多数あり)をメールでタイムリーに配信してくれます。業界の全員がPraise-Netの会員といっても過言ではないです。
| Q17:【市販後】【治験】 市販後副作用等報告において、30 日以内の報告の対象であると考えていたところ、 第一報を報告する前に追加情報により 15 日以内の報告の対象であることが判明した場合の報告期限はいつか? また、治験副作用等報告において、15 日以内の報告の対象であると考えていたところ、第一報を報告する前に追加情報により 7 日以内の報告の対象であることが判明した 場合の報告期限はいつか? | A17: 【市販後】 15日以内の報告の対象であることが判明した日を起算日として15日以内に報告すること。ただし、この報告期限が30日以内の報告の対象であると考えた情報を入手した日を起算日として30日を超える場合は、少なくとも、30日以内の報告の対象であると 考えた情報を、その情報を入手した日を起算日とした30日以内に報告すること。 【治験】 7日以内の報告の対象であることが判明した日を起算日として7日以内に報告する こと。ただし、この報告期限が15日以内の報告の対象であると考えた情報を入手した 日を起算日として15日を超える場合は、少なくとも、15日以内の報告の対象であると考えた情報を、その情報を入手した日を起算日とした15日以内に報告すること。 |
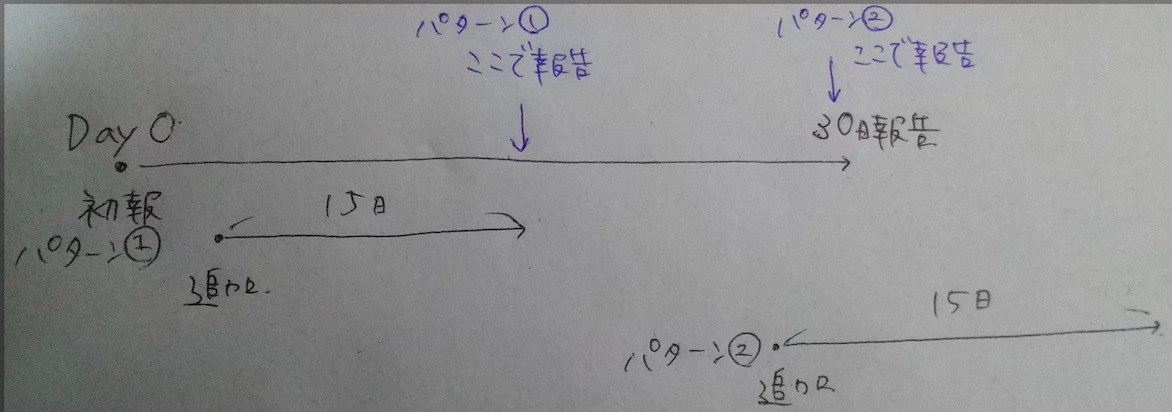 汚い絵でごめんなさい…追加で入手した情報の期日を計算した上で、より報告日が直近で設定されている方を選択する。
汚い絵でごめんなさい…追加で入手した情報の期日を計算した上で、より報告日が直近で設定されている方を選択する。
パターン①は第一報受けて直ぐに追加情報を入手したから15日の期日が直近だからそっちに変更。
パターン②は30日手前に追加情報を入手したら、元々の期日が直近になるからそのまま。
| Q18:【治験】 既に報告済みの症例について、追加の医学的に重要な情報を入手し、追加報告を提出する際には法令上の報告期限は追加の情報入手日を起算日として考えてよいか? | A18:【治験】 よい。追加の情報入手日を起算日として期限内に追加報告すること。例えば、既に15 日以内の報告を行った症例について、7日以内の報告対象となる情報を追加で入手した場合には、7日以内に追加報告を行うこと。なお、既に7日以内に報告済の症例について、報告対象となる情報を追加で入手した場合、追加情報をもって症例の報告期限が15日以内に変更されない限り、7日以内に追加報告を行うこと。 |
補足:外資系企業あるあるです。上司が外国人であまり日本の規制を理解していなかったら注意してください。
海外だと、基本は初回+死亡・生命危機=7日で報告。
イギリスで死亡の追加情報は+8日ルールがあったりなかったりです。
でも、日本は一番期日がタイトで、一度7日報告としたらずーーーっと7日報告。追加報告で実は「15日報告」だった、「報告対象外」、「取下げ」でない限りずっと7日報告。
海外だと、基本は初回+死亡・生命危機=7日で報告。
イギリスで死亡の追加情報は+8日ルールがあったりなかったりです。
でも、日本は一番期日がタイトで、一度7日報告としたらずーーーっと7日報告。追加報告で実は「15日報告」だった、「報告対象外」、「取下げ」でない限りずっと7日報告。
| Q19:【治験】 治験国内管理人が治験副作用等報告を行うにあたり、報告期限はどのように設定すべきか? | A19:【治験】 本邦内に住所を有しない治験の依頼をした者又は治験国内管理人のいずれか早い方が、報告すべき情報を入手した日を日本時間に換算して報告起算日とすることにより 設定すること。 |
補足:治験依頼者が起算日(Day0)の起点になっています。例えば、外国症例って本来本国が入手してそこが起算日となる。
治験国内管理人の場合:本国が入手した日
日本に法人があって、そこが治験依頼者となる:日本が入手した日
つまり日本に法人がある方が起算日を治験国内管理人より後に設定されることを意図しています。
(3)予測性
| Q20:【市販後】 「使用上の注意」の項目のうち予測できるかどうかの判断に用いる項目は何か? | A20:【市販後】 「医療用医薬品の添付文書等の記載要領について」(平成29年6月8日付け薬生発 0608第1号)に基づいて記載されている添付文書では、以下の項目が該当する。 「1.警告」、「2.禁忌」、「5.効能又は効果に関連する注意」「7.用法及び用量 に関連する注意」、「8.重要な基本的注意」、「9.特定の背景を有する患者に関する 注意」、「10.相互作用」、「11.副作用」、「12.臨床検査結果に及ぼす影響」、「13.過 量投与」、「14.適用上の注意」 また、「医療用医薬品添付文書の記載要領について」(平成9年4月25日付け薬発第 606号)及び「医療用医薬品の使用上の注意記載要領について」(平成9年4月25日付 け薬発第607号)に基づいて記載されている添付文書では、以下の項目が該当する。 「警告」、「禁忌」、「原則禁忌」、「効能又は効果に関連する使用上の注意」、「用法及 び用量に関連する使用上の注意」、「慎重投与」、「重要な基本的注意」、「相互作用」、 「副作用」、「高齢者への投与」、「妊婦、産婦、授乳婦等への投与」、「小児等への投 与」、「臨床検査結果に及ぼす影響」、「過量投与」、「適用上の注意」 |


コメント